Variation consequence types, such as “intronic” or “non-synonymous”, describe the variation location or effect of a variation on a transcript. For the latest version of Ensembl (release 62) we have made some significant changes to the way in which we determine these consequence types, and we’d like to provide an overview of these improvements.
Firstly, we are now able to assign a specific effect to every allele of a variant. For example, rs12795274 has three alleles, the reference allele is T, and it also has two alternative alleles; C and A. The A is predicted to cause an amino acid change, while the C is synonymous. We now list the effect of each individual allele on the website and you also can fetch them separately when using the variation API
Another improvement we’ve made is that “under the hood” we now use terms defined in the Sequence Ontology (SO) to describe the consequence types. Moving to this set of externally maintained terms should make it easier to compare Ensembl annotations with those from other groups. The SO also groups the various terms we use into a hierarchical tree and, in the future, this will let users query for variants with particular effects in a much smarter way than is possible now. On the website we are still using our old terms by default, but you can see the mapping between the old terms and the SO terms on the variation documentation page and you can use “Configure this page” on several variation views to choose which set of terms you want to see (here‘s an example).
We also now provide SIFT and PolyPhen predictions for any variant that is predicted to cause an amino acid substitution in human. These are popular tools developed by external groups that try to predict the effect of a non-synonymous mutation on the function of the protein. You can see these predictions on several variation views, a useful example is the protein variation view. You can find more information about these tools and how we run them in Ensembl on the variation documentation page.
![CropperCapture[1402]](https://www.ensembl.info/wp-content/uploads/2011/04/CropperCapture1402.jpg)
All of these improvements are also available for you to use to analyse your own data using the Variant Effect Predictor (VEP). The VEP has new configuration options that allow you to choose which set of terms you want to use for the consequence annotations, and also offers options to fetch SIFT and PolyPhen predictions for any missense mutations in your data. We are able to provide these predictions for novel mutations by computing the predictions from SIFT and PolyPhen for all possible amino acid substitutions in human proteins and storing these in the variation database. We hope that this makes the VEP even more useful for mining your data and we have plans to add support for these sort of tools in other species in the near future.

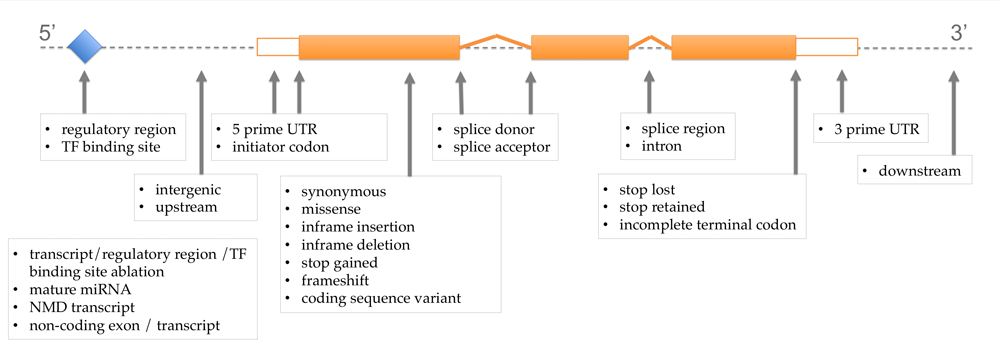 As you will see, the SO equivalents for our old terms are fairly straightforward. The most notable difference is that we have replaced “non-synonymous” with the more specific term “missense”, for changes in amino acid which do not include stop gained, as we already have a specific term for stop gained.
As you will see, the SO equivalents for our old terms are fairly straightforward. The most notable difference is that we have replaced “non-synonymous” with the more specific term “missense”, for changes in amino acid which do not include stop gained, as we already have a specific term for stop gained.


